
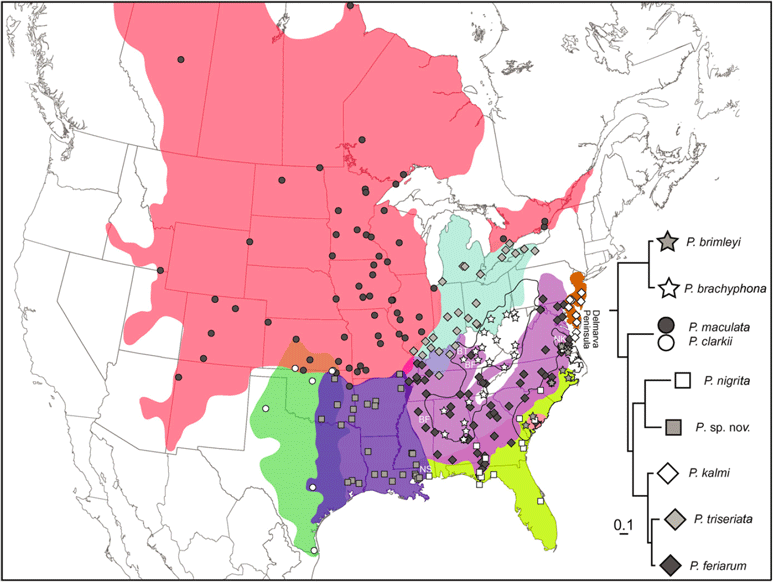
A phylogeny is a bifurcating tree defining the evolutionary relationships among a group of individuals either within (i.e. a genealogy) or among (i.e. a species tree) species. Phylogenies, which can be estimated using genetic or morphological data, serve as the foundation for many downstream evolutionary analyses. Accuracy in phylogeny estimation, therefore, is of critical importance. We focus on two areas of phylogentics 1) large scale phylogeny estimation using genetic data, and 2) identifying sources of error in phylogeny estimation. In the first area we have developed a novel application of the genetic algorithm (GA), the "metapopulation genetic algorithm" that utilizes population structure, along with standard GA components (mutation, selection, recombination) to dramatically improve the efficiency with which large phylogenies can be estimated. We are currently looking for novel applications of this powerful algorithm. In the second area we have used carefully-constsructed simulations to test the effects of model-misspecification and missing data on the accuracy of phylogeny estimates.