
One of the fundamental questions in evolutionary biology is: How do new species arise? Empirical biologists have postulated that barriers to gene flow develop through geological processes, such as the formation of rivers and mountains, and through climatic processes, such as the formation of glaciers and deserts. Biologists often encounter difficulties, however, when trying to identify the specific processes driving speciation in a particular group. These difficulties result, in part, from the lack of a statistical framework for integrating the diverse types of data needed to study historical patterns of speciation.
In recent years, there has been an explosive increase in the amount of phylogenetic, ecological, and geological data available to biologists studying speciation. Phylogenetic data are becoming available for a growing number of taxonomic groups and at an increasingly continuous geographic scale. Ecological data, in combination with sampling localities from museum databases, are being used to generate estimates of species’ ranges and the ecological parameters that limit these ranges. Geological data are providing ever-more precise spatial locations of the types of features (e.g. rivers) that are expected to restrict gene flow and contribute to speciation.
While data from these diverse fields are accumulating rapidly, current phylogeographic methods cannot integrate this information (Knowles and Maddison, 2002). Some methods lack a statistical framework, making it difficult to test explicit hypotheses in a rigorous manner. Other methods make very restrictive geographic assumptions or are computationally intractable for large data sets. What is needed, therefore, is a spatially explicit and computational tractable statistical framework that can make use of the geographically continuous data resulting from phylogenetics, ecology, and geology.
PhyloMapper implements a likelihood-based statistical framework for estimating historical patterns of gene flow and ancestral geographic locations. This method uses a phylogeny with branch lengths and the geographic localities of all individuals represented by the tips of the phylogeny. Using a spatially-explicit model of migration related to the diffusion model, I have derived an equation describing the likelihood of observing the haplotypes sampled at their current geographic locations, given the locations of their ancestors and the dispersal distance of the species. Using a maximum likelihood approach, Emily Moriarty Lemmon and I have recently applied this new framework to a 246-taxon phylogeny of chorus frogs (Pseudacris; Lemmon et al. 2007) and tested hypotheses regarding phylogeographic structure, routes of expansion, and glacial refugia (Lemmon and Lemmon, 2008). This framework, which represents an important advance in the field of phylogeography, has been implemented in the Java software PhyloMapper (available at the download site).
Future Directions...
(click figure to enlarge)
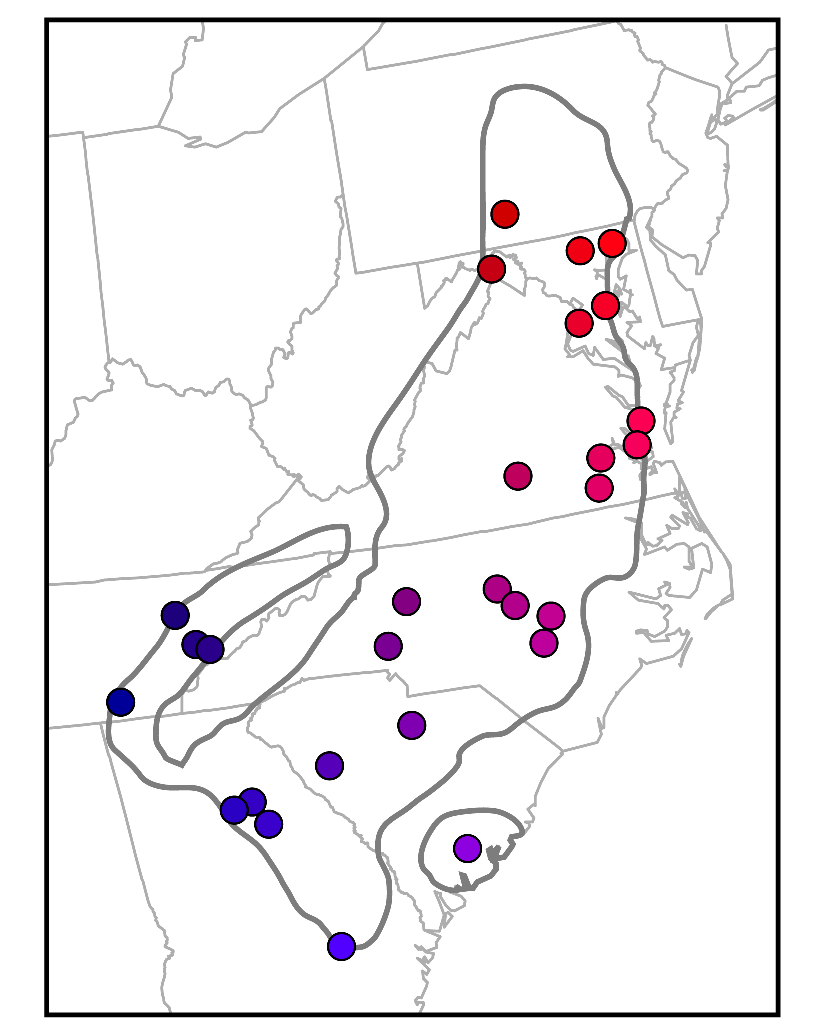
Data I: Geographic Coordinates
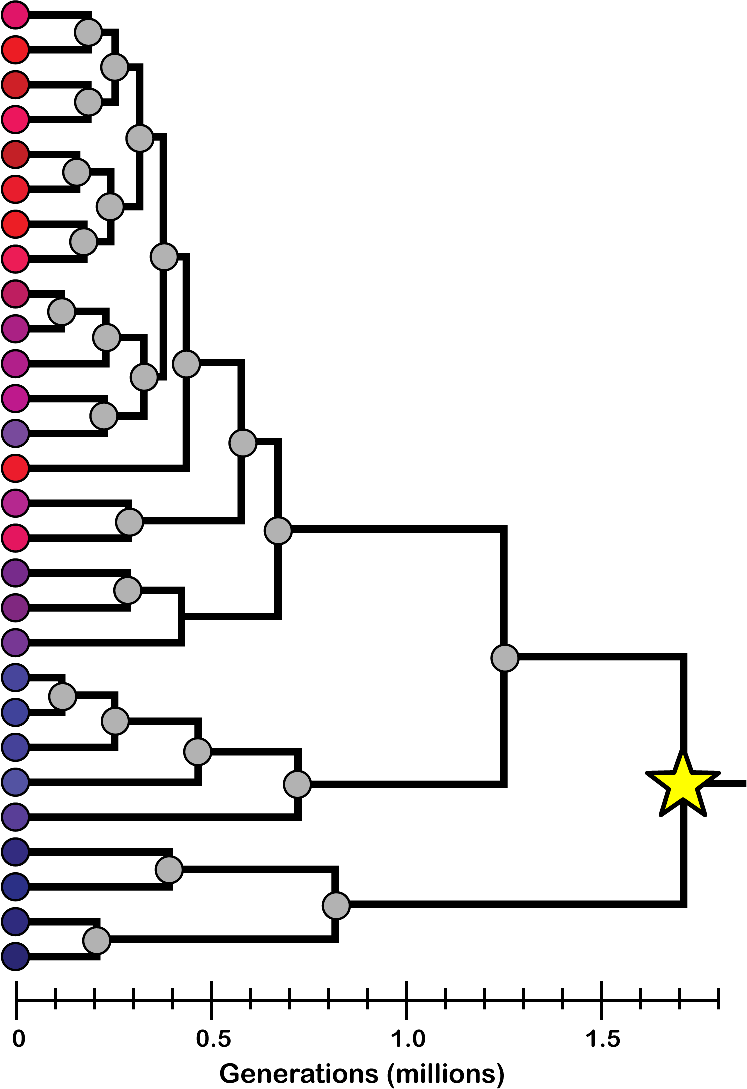
Data II: Genealogy
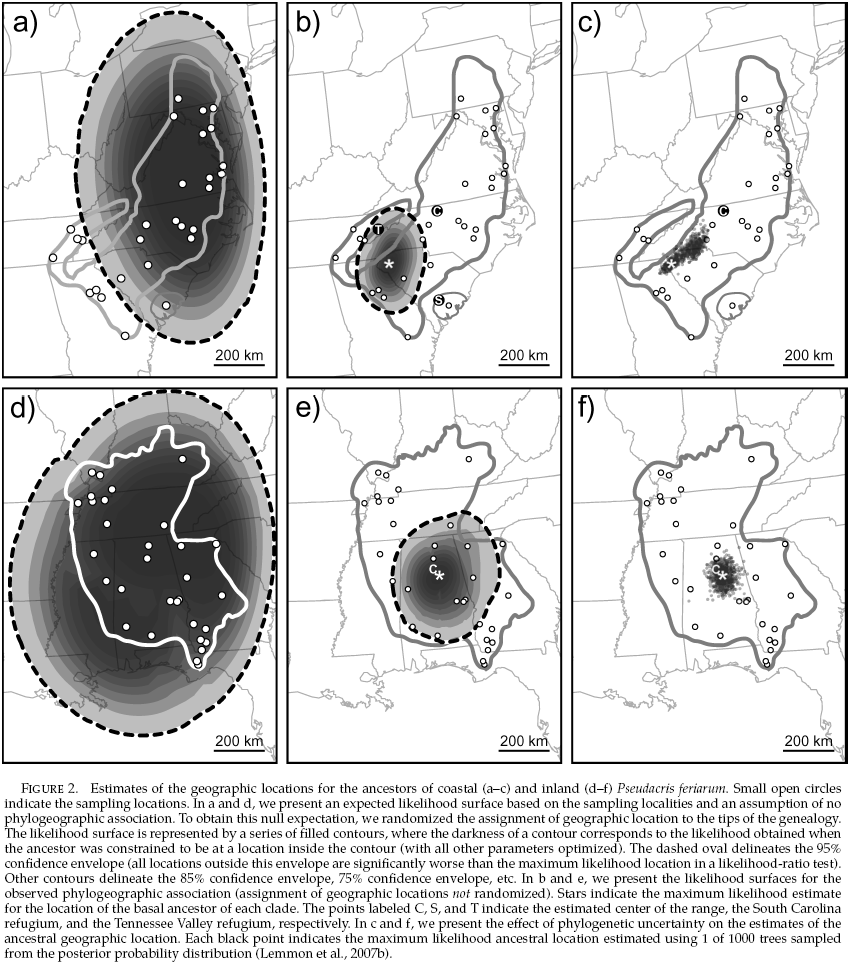
Ancestral Location Estimate
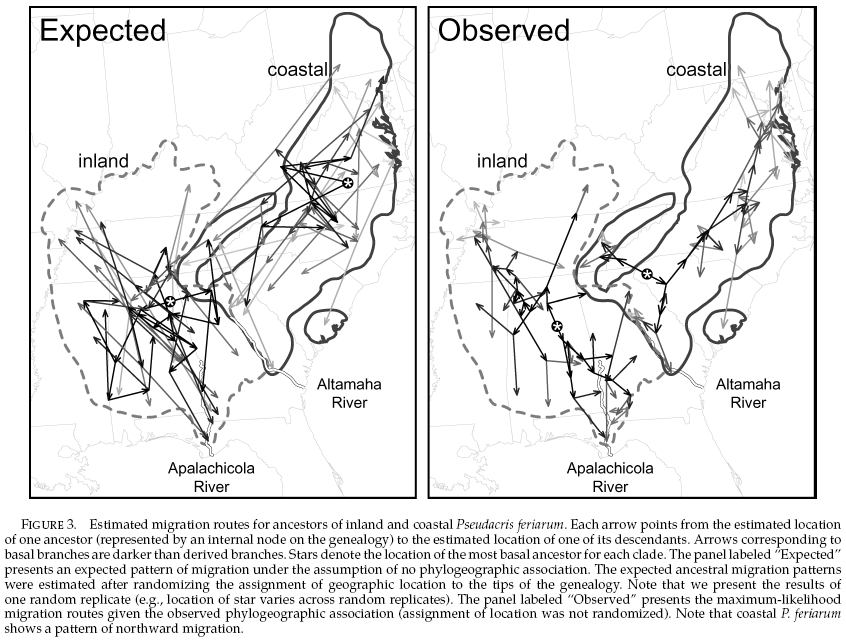
Dispersal Patterns Estimate
(released September 29, 2008)